Konference: 2011 XVIII. Jihočeské onkologické dny
Kategorie: Kolorektální karcinom
Téma: Varia
Číslo abstraktu: 30
Autoři: MUDr. Helena Štěpánková; MUDr. Anna Křepelová; Doc.MUDr. Lenka Foretová , Ph.D.
Hereditární nepolypózní kolorektární karcinom (HNPCC, Lynchův syndrom) patří mezi hereditární nádorové syndromy s vysokou predispozicí ke kolorektálnímu karcinomu (CRC). Na podkladě tohoto syndromu vzniká cca 2-5 % CRC.
Dispozice je daná mutacemi v tzv. mutátorových genech (MMR geny), především MSH2, MLH1, MSH6. Jedná se o geny identifikující a opravující chyby při replikaci DNA. Zárodečná mutace je u těchto jedinců přítomna ve všech somatických buňkách, k iniciaci vzniku tumoru stačí mutace/eliminace druhé alely genu (ztráta heterozygozyty), důsledkem je nefunkční nebo žádný produkt a zahájení mnohastupňového procesu akumulace poruch základních regulačních mechanismů buněk. Dědičnost dispozice je autosomálně dominantní s 50% rizikem přenosu na potomky bez rozdílu pohlaví. Celoživotní riziko pro CRC je až 80%, dále je vysoké riziko i extrakolické lokalizace nádorů - endometria, ureteru, ledvinné pánvičky, tenkého střeva, hepato-biliárního traktu... Penetrance (klinický projev) je variabilní, ovlivněna i negenetickými faktory a pohlavím.
Kazuistika:
Na kazuistice je prezentována problematika diagnostiky, dispenzarizace, presymptomatického či prenatálního vyšetření.
Obr. 1. Probandkou v rodině je žena, která onemocněla CRC ve 23 letech, kolonoskopie byla provedena po roce GIT obtíží i při významné rodinné anamnéze (RA) až na její žádost. V rodině nebyla splněna Amsterodamská kritéria k DNA vyšetření, molekulárně genetické vyšetření pro susp. HNPCC bylo zahájeno vzhledem k nízkému věku a RA. Sekvenační analýza genů MLH1 a MSH2 neodhalila kauzální mutaci.
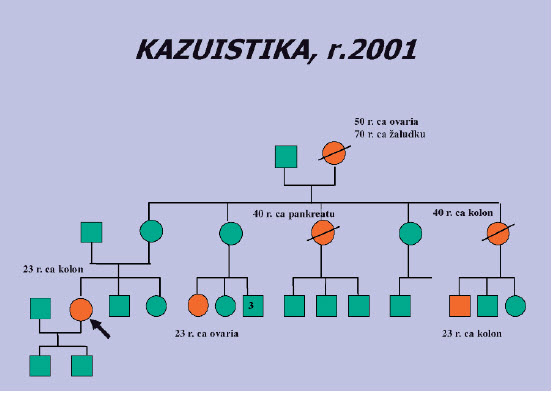
Obr. 2. Po zavedení nové vyšetřovací metody (MLPA) byla po 3 letech nalezena delece exonů 1 -2 v genu MSH2 a tím potvrzen Lynchův syndrom. Během té doby však zemřela matka probandky na CRC, syn onemocněl ve 4 letech anaplastickým astrocytomem. Před jeho úmrtím matka nedala souhlas k DNA vyšetření a tak nebyla objasněna možná souvislost s familiární dispozicí - CRC + nádor mozku je označován jako Turcotův syndrom.
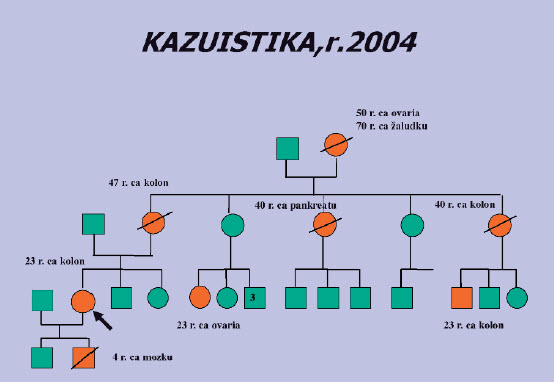
Obr. 3. V dalším roce byla pacientka odeslána gynekologem v 17. týdnu gravidity k prenatální diagnostice Lynchova syndromu. U tohoto dědičného nádorového syndromu je možné v případě nálezu kauzální mutace nabídnout rodině preimplantační diagnostiku, prenatální diagnostika se neprovádí, je možné ji zvážit výjimečně v případě rizika homozygozyty (zátěž ze strany obou rodičů) a rizikem malignit již v dětském věku (CRC, hematologické malignity, neuroblastom).
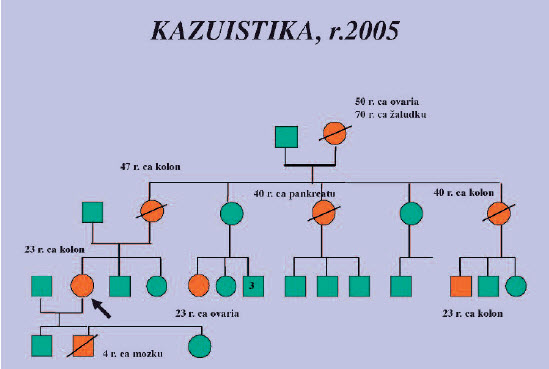
Rodina byla informována prostřednictvím probandky a nabídnuto vyšetření jak nemocných, tak prediktivní vyšetření příbuzných v riziku. V současné době dle informací probandky je část rodiny sledovaná, sestra se nyní rozhoduje podstoupit molekulárně genetické vyšetření.
Závěr:
K vyhledávání rodin s vysokým rizikem nádorového onemocnění je nezbytný aktivní přístup, zohlednění rodinné anamnézy (orgánové postižení, věk výskytu, metachronní či synchronní tumory, histologická specifikace aj.).
Presymptomatické (prediktivní) DNA vyšetření se u zdravých příbuzných v riziku provádí od 18 let, v případě onemocnění v dětském věku se souhlasem zákonného zástupce.
Rodinám s nálezem kauzální mutace lze nabídnout v případě plánované gravidity preimplantační diagnostiku. DNA vyšetření indikuje klinický genetik po provedení genetické konzultace a s informovaným souhlasem vyšetřovaného.
Negativní molekulárně genetické vyšetření v rodinách se zvýšeným výskytem nádorových onemocnění neznamená vyloučení hereditárního rizika, i v těchto rodinách je nezbytné sledování dle empirického rizika.
Literatura:
-
Plevová P., Novotný J., Šachlová M., Křepelová A., Foretová L. Hereditární nepolypózní kolorektální karcinom. Klinická onkologie, Supplementum 2009; 12-15.
-
Goetz P., Krutílková V. Kancerogeneze. Postgrad. Medicína 2002; 544-551.
-
Křepelová A., Pavlíková K., Plevová P. Diagnostika Lynchova syndromu - nové geny a metody. Klinická onkologie, Supplementum 2006; 76-81.
-
Zikán M., Sláma J., Pinkavová I., Fischerová D., Freitag P., Cibula D. Dědičná dispozice ke vzniku karcinomu endometria, Čes. gynekologie 2011, 76. Č. 3; 176-179.
-
Vasen HFA., Môslein G., Alonso A. et al. Guidelines fothe clinical management of Lynch syndrome (hereditary non-polyposis cancer). J. Med genet 2007; 44; 353-362.
-
Húttelová R., Kleibl Z., Řezáčová J., Krutílková V., Foretová L., Novotný J., Kotlas J., Zikán M., Pohlreich P. Předpoklady pro neimplantační genetickou diagnostiku (PGD) u nosičů mutací v nádorových predispozičních genech. Klinická onkologie; 22, Supplementum 2009, 69-74.
-
Baxová A., Martásek P. Komentář: preimplantační genetická diagnostika. Klinická onkologie; 22, Supplementum 2009, 75.
-
Goetz P., Floretová L., Puchmajerová A. Hereditární etiologie nádorových onemocnění a význam genetického poradenství a testování v onkologii. Klinická onkologie, Supplementunt 2006; 44-47.
-
Databáze Online Mendelian Inheritance in Man (OMIM).
Datum přednesení příspěvku: 15. 10. 2011