Konference: 2015 20th Congress of the European Hematology Association - účast ČR
Kategorie: Maligní lymfomy a leukémie
Téma: CLL - Biology: Interacting determinants of CLL ontogeny and evolution
Číslo abstraktu: S121
Autoři: Viktor Ljungström; Diego Cortese; Emma Young; Tatjana Pandzic; Larry Mansouri; Mgr. Karla Plevová, Ph.D.; Lesley-Ann Sutton; Niki Stavroyianni; Andreas Agathangelidis; M.D. Davide Rossi; M.D. Martin Hoglund, Ph.D.; Prof. David Graham Oscier; MD Gianluca Gaidano, PhD; MD Frederic Davi, PhD; Dr. Christianne Pott, PhD; Dr. Livio Trentin; prof. RNDr. Šárka Pospíšilová, Ph.D.; M.D. Paolo Ghia, Ph.D.; MD Kostas Stamatopoulos; Tobias Sjöblom; Prof. MD Richard Rosenquist (Brandell), PhD
Background
Fludarabine, cyclophosphamide and rituximab (FCR) is the
gold-standard first-line regimen in medically fit patients with
chronic lymphocytic leukemia (CLL); however, despite good response
rates most patients will eventually relapse. BesidesTP53
aberrations, the mechanisms leading to relapse after FCR treatment
are currently poorly understood.
Aims
To characterize the genetic mechanisms underlying relapse following
treatment with FCR using whole-exome sequencing (WES).
Methods
Forty-one CLL patients receiving FCR with either a partial response
(PR, with ≥4 cycles of treatment completed) or a complete response
(CR, ≥1 cycle of treatment completed) were selected. Pre-treatment
and relapse samples (mean time to relapse 3.2 years, range 0.7 –
10.9), together with matched germline DNA for 28 patients, were
analyzed by WES. Well-established bioinformatics tools and
pipelines were used to process raw sequencing reads, enabling the
identification of somatic mutations and also facilitating the
analysis of copy-number aberrations (CNA) and absolute cancer cell
fractions (CCF).
Results
Amongst the 28 patients with matched germline DNA, 1191 somatic
variants (>10% allele frequency) were found in the pre-treatment
samples and 1334 in the relapse samples, with an average of 15.2
(range, 3-24) and 17.6 (range, 2-32) non-silent mutations per case,
respectively. Mutations were predominantly missense substitutions
(81%) and less frequently frameshift or in-frame
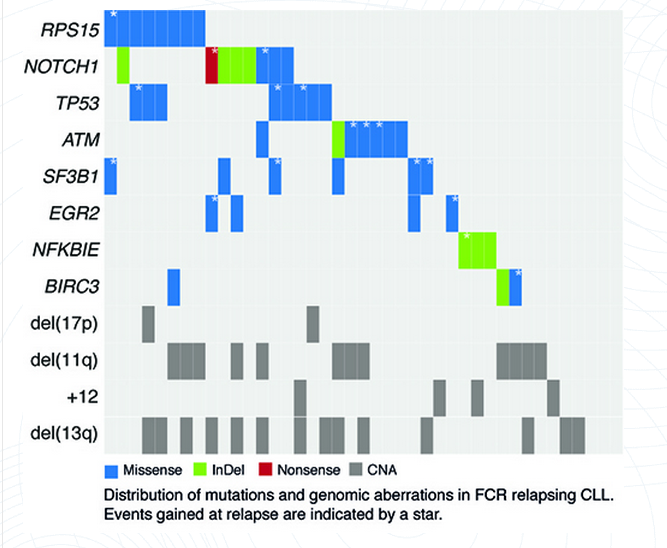
insertions/deletions (14%) or nonsense mutations (5%). As expected,
at relapse, a high proportion of cases harbored mutations in genes
previously linked to adverse prognosis in CLL: TP53 (n=8;
19.5%), NOTCH1 (n=8; 19.5%), ATM (n=7; 17%),
SF3B1 (n=6; 14.6%), NFKBIE (n=4; 9.8%),
EGR2 (n=4; 9.8%) and BIRC3 (n=3; 7.3%).
Intriguingly, a large proportion of cases also harbored mutations
in RPS15 (n=8; 19.5%), a gene encoding a component of the
40S ribosomal subunit. High allele frequencies were observed for
RPS15 mutations at both time points (range, 29% - 56%),
and all mutations were missense variants residing within a 7
amino-acid evolutionarily conserved region. Besides its role in
protein translation, RPS15 has been shown to stabilize p53 by
interfering with the MDM2-p53-MDMX network and inhibiting
MDM2-mediated p53 degradation. Characterization of two
recurrent RPS15 mutations in the HCT116 colorectal cancer
cell line transiently expressing either wild-type (wt) or mutant
RPS15 revealed impaired ability of RPS15P131S and
RPS15G132A in regulating endogenous p53. As both
mutations map within the region that interacts with MDM2, this
finding strongly suggests that binding of RPS15P131S and
RPS15G132A to MDM2 is less efficient compared to wt
protein thus leading to more pronounced p53 degradation. Finally,
by calculating the absolute CCF for all mutations at both time
points allowed monitoring of clonal heterogeneity over time. All 24
cases with available exome-derived CNA data showed mutations
expanding ≥0.3 in CCF between the time points (mean 7.4 mutations,
range 1-21). Among recurrently mutated genes, i)RPS15
remained stable over time, ii) TP53, EGR2,
NOTCH1 and BIRC3 mutations expanded or remained
stable, and iii) forSF3B1 and ATM mutations
both increasing and decreasing CCFs were observed.
Summary
We provide novel insights into the heterogeneous genetic landscape
of CLL relapsing after FCR treatment with our most prominent
finding being recurrent RPS15 mutations (19.5%) and with
in vitro studies of RPS15 mutations pointing to a
novel mechanism for p53 dysregulation in CLL.
Keyword(s): Chronic lymphocytic leukemia, Mutation
analysis
Datum přednesení příspěvku: 12. 6. 2015